Biomarqueurs diagnostiques liés au lysosome | JIR

Introduction
La prévalence du sepsis pédiatrique, dont les causes sont identifiées ou non, constitue une menace importante pour le bien-être et la survie d’environ 3 millions d’enfants chaque année.1 Malgré le risque de surconsommation d’antibiotiques, l’antibiothérapie empirique a démontré son efficacité dans la gestion de l’infection, la stabilisation de l’hémodynamique et la modulation de la réponse septique.2 Malheureusement, le sepsis pédiatrique présente une hétérogénéité substantielle tout au long du processus pathobiologique, ce qui pose des défis importants aux cliniciens dans la prise de décisions appropriées, telles que l’administration d’antibiotiques au stade précoce et le traitement de soutien organique, ainsi que l’évaluation des risques et l’arrêt de l’utilisation d’antibiotiques au stade tardif du sepsis.3 Par conséquent, pour diagnostiquer, traiter et prévenir efficacement le sepsis pédiatrique, il est impératif d’explorer activement les biomarqueurs qui peuvent déterminer avec précision le sepsis pédiatrique.
Les lysosomes, qui sont des organites membranaires responsables de la dégradation des macromolécules intracellulaires, jouent un rôle crucial en tant que centres de signalisation pour la régulation du calcium et le contrôle de la réponse aux nutriments. Par conséquent, la préservation de l’intégrité et de la fonctionnalité des lysosomes est vitale pour le maintien de l’homéostasie cellulaire.4 On sait que les lysosomes jouent un rôle important dans l’inflammation, la présentation des antigènes et la préservation des cellules immunitaires à longue durée de vie.5 Divers types de stress peuvent déclencher une augmentation de la perméabilité de la membrane lysosomale, entraînant la décharge de composants intracellulaires des lysosomes dans le cytoplasme et conduisant finalement à la mort cellulaire qui dépend des lysosomes.6 L’activation excessive de l’inflammasome NLRP3 peut induire un clivage lysosomal, qui joue un rôle dans la régulation de l’inflammation au cours de la septicémie.7 Les pièges extracellulaires des neutrophiles (NET) peuvent favoriser la libération de la cathepsine B (CatB) à partir de lysosomes rompus en induisant une apoptose thermique dans les macrophages (Mϕ) et en modulant ainsi la réponse inflammatoire en cas de septicémie.8 Le complexe Ragulator, fixé à la membrane lysosomale, peut participer à la progression des maladies associées à l’inflammation en activant l’inflammasome NLRP3 intracellulaire.9 Dans un environnement lysosomal acide, HMGB1 peut induire une fuite de LPS dans le cytoplasme, provoquant des lésions pulmonaires aiguës et la mort des enfants.10 Les effets de la morphine sur les processus lysosomaux, tels que la production, l’acidification et la fusion mitochondriale-lysosomale, entraînent une diminution de l’activation de l’autophagie mitochondriale dans la microglie, ce qui favorise finalement le choc infectieux.11 La mélatonine, associée à une modulation du stress anti-oxydatif et de l’autophagie lysosomale, réduit la survenue d’un dysfonctionnement multiorganique associé au sepsis.12 Les α-MOF peuvent libérer des ions calcium et zinc dans les lysosomes des monocytes/macrophages pour favoriser la dégradation bactérienne et améliorer efficacement la survie des patients septiques.13 Le ciblage des lysosomes est donc une stratégie thérapeutique potentielle pour moduler l’inflammation et le métabolisme dans diverses conditions.
Dans cette étude, nous avons intégré de multiples ensembles de données de séquençage à haut débit de la septicémie pédiatrique pour développer un modèle de diagnostic axé sur la fonction lysosomale, qui identifie efficacement les cas de septicémie pédiatrique. La fiabilité de ce modèle de diagnostic a ensuite été vérifiée à l’aide d’une cohorte comprenant 30 individus sains et 35 enfants diagnostiqués avec une septicémie, sur la base d’échantillons de sang périphérique. Ce modèle de diagnostic devrait offrir aux professionnels de la santé de nouvelles perspectives en matière de diagnostic et de traitement de la septicémie pédiatrique.
Matériel et méthodes
Acquisition des cohortes de septicémie pédiatrique et des gènes lysosomiaux
Nous avons conçu l’étude actuelle conformément au Transparent Reporting of Individual Prognostic or Diagnostic Multivariate Predictive Models (TRIPOD). Trois ensembles de données (GSE13904, GSE26378 et GSE26440) comprenant des profils d’expression génique du sang total d’enfants atteints de septicémie et de témoins sains ont été acquis à partir de la base de données gene expression omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo/). La normalisation de chaque ensemble de données a été effectuée à l’aide de la fonction “normalizeBetweenArrays” du paquet R “Limma”. Les effets de lot entre les trois ensembles de données ont été corrigés à l’aide de la fonction Combat du package R “SVA”. GSE26378 (21 sepsis normaux contre 82 sepsis pédiatriques) a servi d’ensemble d’apprentissage pour l’analyse ultérieure, tandis que GSE26440 (32 sepsis normaux contre 98 sepsis pédiatriques) et GSE13904 (18 sepsis normaux contre 106 sepsis pédiatriques) ont été utilisés en tant qu’ensembles de validation. En outre, un ensemble complet de 894 gènes associés aux lysosomes a été obtenu à partir de la base de données Gene Ontology (GO) (https://geneontology.org/) et la base de données Gene Set Enrichment Analysis (GSEA) (http://www.gsea-msigdb.org/).
Criblage des gènes différentiellement exprimés (DEG)
Les gènes différentiellement exprimés ont été identifiés entre les échantillons de sepsis pédiatriques et les échantillons sains dans l’ensemble de données de formation, en utilisant des valeurs P ajustées inférieures à 0,05 et |logFC| > ; 1 comme critères. La base de données STRING (http://string-db.org) a été utilisée pour effectuer une analyse du réseau d’interactions protéine-protéine (PPI) sur la base de ces DEG. Par la suite, une analyse d’enrichissement de l’ensemble des gènes (GSEA)14 a été utilisé pour identifier les voies de signalisation enrichies par les DEG.
Développement de biomarqueurs diagnostiques liés aux lysosomes
Les gènes chevauchants des DEG et les gènes lysosomaux dans la septicémie pédiatrique ont été soumis à une analyse plus poussée dans l’ensemble de données GSE26378. Dans un premier temps, une analyse de régression LASSO (least absolute shrinkage and selection operator) a été réalisée à l’aide du package R “glmnet” afin de sélectionner les paramètres optimaux par validation croisée 10 fois. Ensuite, une analyse de forêt aléatoire a été réalisée à l’aide du paquetage R “randomForest” pour filtrer les paramètres optimaux. Les données de sortie de caisse associées ont été utilisées pour déterminer l’erreur de sortie de caisse de chaque arbre de décision. ErrorOOB1 peut être utilisé pour représenter la valeur de l’erreur. Pour déterminer l’erreur de données hors poche errorOOB2, le bruit a été ajouté au hasard aux données hors poche I. La pertinence des données hors poche I (score(I)) a finalement été calculée à l’aide de la formule suivante 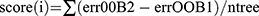 où ntree est le nombre d’arbres de décision. Le nombre d’arbres de décision à utiliser a été choisi entre 100 et 1000, et les valeurs par défaut ont été utilisées pour tous les autres paramètres afin d’ordonner l’importance des caractéristiques. En outre, les gènes partagés dans les deux algorithmes d’apprentissage automatique ont été sélectionnés pour l’analyse de régression logistique afin de développer un modèle de diagnostic pour la septicémie pédiatrique. Par la suite, la validité du modèle de diagnostic a été évaluée à l’aide de l’analyse de la courbe caractéristique d’exploitation du récepteur (ROC) dans les ensembles de données d’apprentissage et de test, et l’aire sous la courbe (AUC) a été calculée pour évaluer la performance prédictive de l’algorithme.
où ntree est le nombre d’arbres de décision. Le nombre d’arbres de décision à utiliser a été choisi entre 100 et 1000, et les valeurs par défaut ont été utilisées pour tous les autres paramètres afin d’ordonner l’importance des caractéristiques. En outre, les gènes partagés dans les deux algorithmes d’apprentissage automatique ont été sélectionnés pour l’analyse de régression logistique afin de développer un modèle de diagnostic pour la septicémie pédiatrique. Par la suite, la validité du modèle de diagnostic a été évaluée à l’aide de l’analyse de la courbe caractéristique d’exploitation du récepteur (ROC) dans les ensembles de données d’apprentissage et de test, et l’aire sous la courbe (AUC) a été calculée pour évaluer la performance prédictive de l’algorithme.
Extraction de l’ARN et analyse par qRT-PCR
Cette étude a porté sur un total de 35 cas de septicémie pédiatrique répondant aux critères diagnostiques de la septicémie,2,15 ainsi que 30 enfants normaux, entre le 1er janvier 2023 et le 30 mai 2023. La confirmation des échantillons de septicémie pédiatrique était basée sur les résultats de l’hémoculture. Les caractéristiques cliniques de base des deux groupes peuvent être consultées à l’adresse suivante Tableau S1. Le groupe septique présentait des niveaux plus élevés de procalcitonine (PCT) et de pourcentage de neutrophiles (NEU%) par rapport au groupe normal. Cependant, aucune différence statistiquement significative n’a été observée en termes d’âge, de sexe, de numération des globules blancs et de taux de protéine C-réactive (CRP). Trois millilitres de sang total ont été prélevés sur chaque enfant après un jeûne. Les cellules mononucléaires du sang périphérique (PBMC) ont été isolées à l’aide d’un isolat de Ficoll-Paque. L’ARN total a été extrait des PBMC et les niveaux d’ARNm des marqueurs diagnostiques ont été détectés par qRT-PCR. La normalisation a été effectuée à l’aide de la β-ACTIN.16 Les séquences d’amorces se trouvent à l’adresse suivante Tableau S2.
Évaluation de l’infiltration des cellules immunitaires
Compte tenu du rôle important des cellules immunitaires dans la pathogenèse de la septicémie pédiatrique,17 le CIBERSORT18 a été utilisée pour évaluer l’infiltration de divers types de cellules immunitaires. Cette analyse visait à élucider les types de cellules immunitaires qui présentent une forte corrélation avec les scores du modèle dans différentes cohortes d’enfants atteints de septicémie.
Analyse statistique
Les données catégorielles ont été analysées à l’aide du test exact de Fisher ou du test du chi carré, tandis que les variables quantitatives ont été évaluées à l’aide de la méthode des échantillons indépendants. t-test t. Toutes les valeurs p ont été considérées comme statistiquement significatives à un niveau de signification de P<0,05, avec une approche bilatérale.
Résultats
Criblage des DEG liés aux lysosomes
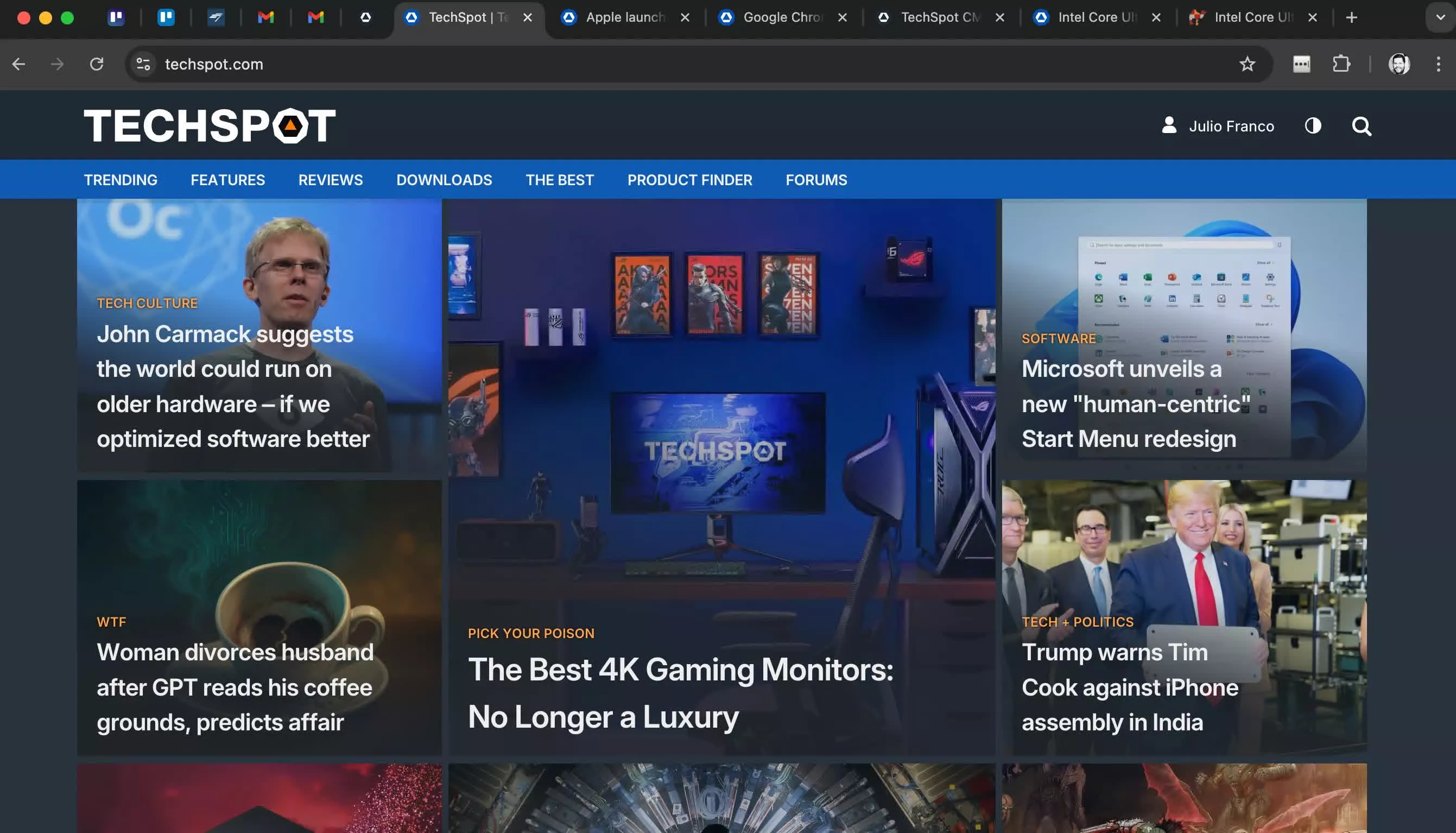
Dans l’ensemble de données GSE26378 (Figure 1A), un total de 468 DEGs régulés à la hausse et 271 DEGs régulés à la baisse ont été identifiés. Par la suite, 83 DEG liés aux lysosomes ont été sélectionnés (Figure 1B). Pour visualiser les interrelations entre les différentes protéines et suggérer des mécanismes potentiels de fonctionnement des protéines, un réseau PPI a été construit à partir de ces DEG associés au lysosome dans la septicémie pédiatrique (Figure 1C). Les résultats de l’analyse GSEA ont indiqué que ces DEG associés aux lysosomes étaient principalement liés à la dérégulation des neutrophiles, aux processus lysosomaux et aux effecteurs immunitaires (Figure 1D).
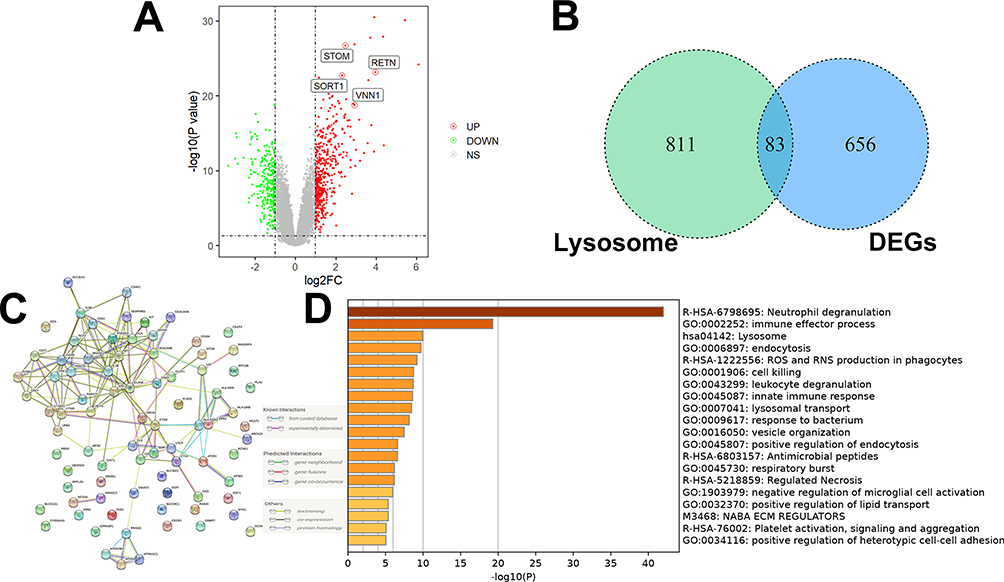 |
Figure 1 Criblage des DEG liés aux lysosomes dans l’ensemble de données GSE26378. (A) Au total, 7468 DEGs régulés à la hausse et 271 DEGs régulés à la baisse ont été identifiés. (B) 83 DEG liés aux lysosomes ont été examinés. (C) Analyse du réseau PPI. (D) Les résultats de l’analyse GSEA ont indiqué que ces DEG associés au lysosome étaient principalement associés à la dérégulation des neutrophiles, aux processus lysosomaux et aux effecteurs immunitaires. |
Construction de marqueurs diagnostiques liés au lysosome
L’analyse de régression LASSO a permis de sélectionner les 13 premiers paramètres (Figure 2A), tandis que l’analyse par forêt aléatoire a également permis d’identifier 13 paramètres optimaux (Figure 2B). Parmi ceux-ci, quatre gènes au total se sont avérés communs aux deux analyses et ont ensuite été utilisés dans le développement d’un modèle de diagnostic de la septicémie pédiatrique (Figure 2C). 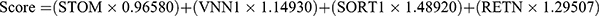 . Par la suite, une analyse ROC a été réalisée sur les cohortes de formation et de validation afin d’évaluer l’efficacité du modèle de diagnostic. Comme le montre la figure Figure 2DLes niveaux d’expression des quatre gènes impliqués dans le modèle de diagnostic étaient systématiquement plus élevés dans le groupe sepsis que dans le groupe normal dans les trois cohortes. Cette tendance a également été observée dans les scores du modèle de diagnostic (Figure 2E). Le modèle de diagnostic a notamment fait preuve d’une capacité de diagnostic remarquable, comme en témoignent les valeurs AUC de 1 dans la cohorte GSE26378, de 0,989 dans la cohorte GSE26440 et de 0,971 dans la cohorte GSE13904, respectivement (figure 2E).Figure 2F).
. Par la suite, une analyse ROC a été réalisée sur les cohortes de formation et de validation afin d’évaluer l’efficacité du modèle de diagnostic. Comme le montre la figure Figure 2DLes niveaux d’expression des quatre gènes impliqués dans le modèle de diagnostic étaient systématiquement plus élevés dans le groupe sepsis que dans le groupe normal dans les trois cohortes. Cette tendance a également été observée dans les scores du modèle de diagnostic (Figure 2E). Le modèle de diagnostic a notamment fait preuve d’une capacité de diagnostic remarquable, comme en témoignent les valeurs AUC de 1 dans la cohorte GSE26378, de 0,989 dans la cohorte GSE26440 et de 0,971 dans la cohorte GSE13904, respectivement (figure 2E).Figure 2F).
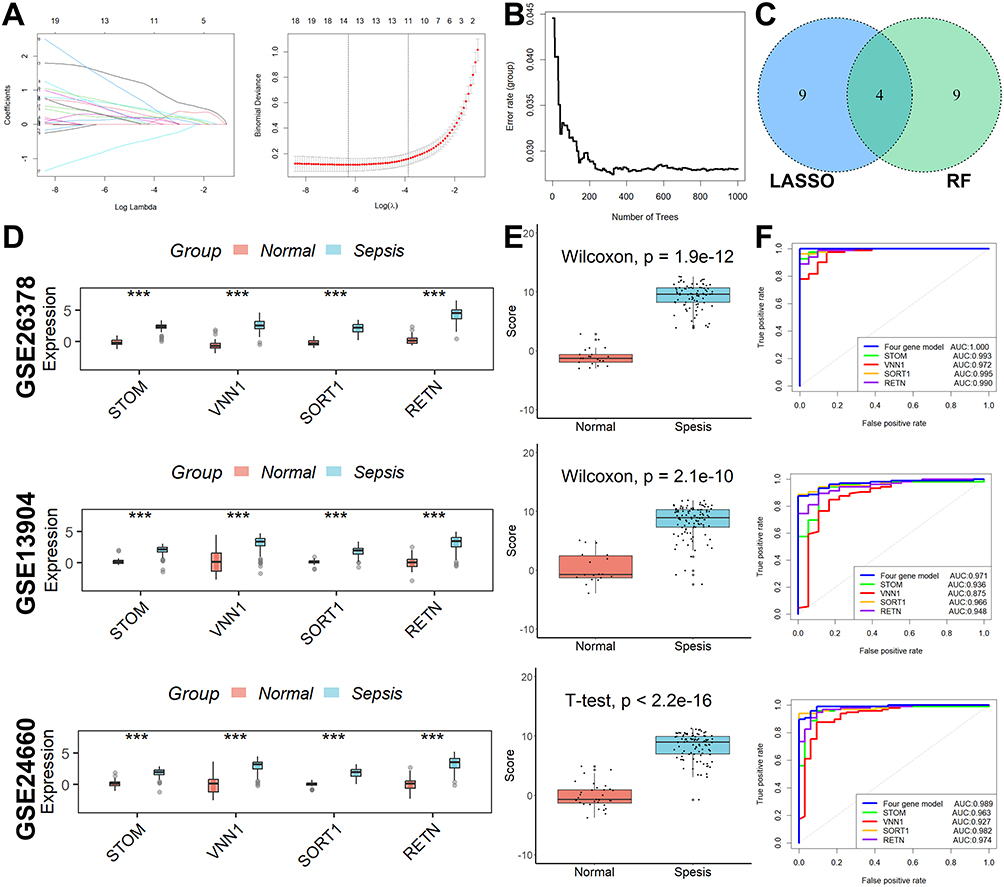 |
Figure 2 Construction et validation de marqueurs diagnostiques liés aux lysosomes. (A) L’analyse de régression LASSO a permis de sélectionner les 13 premiers paramètres. (B) L’analyse de la forêt aléatoire a également permis d’identifier 13 paramètres optimaux. (C) Quatre gènes se chevauchent. (D) L’expression des quatre gènes impliqués dans le modèle de diagnostic était plus élevée dans le groupe septique que dans le groupe normal dans les trois cohortes. (E) Les scores du modèle de diagnostic étaient significativement plus élevés dans le groupe sepsis que dans le groupe normal dans les trois cohortes. (F) Le modèle de diagnostic a montré une bonne capacité de diagnostic. ***p < ; 0,001. |
Analyse des performances des marqueurs diagnostiques dans une cohorte clinique
Les souches bactériennes responsables de la septicémie chez l’enfant sont représentées dans le tableau suivant Figure 3A. Conformément aux résultats de l’analyse bioinformatique susmentionnée, les niveaux d’expression des quatre gènes impliqués dans le modèle de diagnostic étaient significativement plus élevés dans le groupe septique que dans le groupe normal (Figure 3B), ainsi que les scores du modèle de diagnostic (Figure 3C). Le modèle de diagnostic a fait preuve d’une efficacité diagnostique louable, comme en témoignent les valeurs de l’aire sous la courbe (AUC) de 0,917 dans les 65 échantillons cliniques (Figure 3D). En outre, le modèle de diagnostic a démontré une efficacité supérieure à celle des marqueurs inflammatoires conventionnels, notamment la PCT, la CRP, le WBC et le NEU% (Figure 3E). En outre, nous avons évalué la capacité du modèle de diagnostic à différencier le sepsis bactérien du sepsis non bactérien et avons observé qu’il possédait un niveau de compétence modéré (Figure 3F).
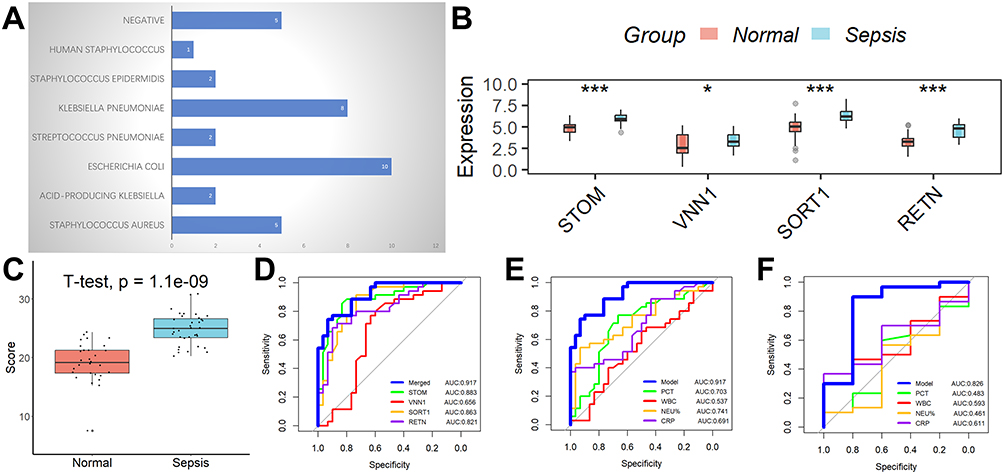 |
Figure 3 Analyse des performances des marqueurs diagnostiques dans une cohorte clinique. (A) Les types de bactéries qui infectent les enfants atteints de septicémie. (B) L’expression des quatre gènes impliqués dans le modèle de diagnostic était plus élevée dans le groupe septique que dans le groupe normal. (C) Les scores du modèle de diagnostic étaient significativement plus élevés dans le groupe sepsis que dans le groupe normal. (D) Le modèle de diagnostic a montré une bonne capacité de diagnostic dans les 65 échantillons cliniques. (E) Le modèle de diagnostic a montré une meilleure efficacité par rapport aux indicateurs inflammatoires conventionnels tels que PCT, CRP, WBC et NEU%. (F) La valeur de ce modèle de diagnostic pour distinguer le sepsis bactérien du sepsis non bactérien. *p < ; 0,05 ; ***p < ; 0,001. |
Analyse de l’infiltration des cellules immunitaires
L’infiltration de divers types de cellules immunitaires dans différentes catégories de septicémie pédiatrique a été évaluée à l’aide de la base de données CIBERSORT. Par la suite, les scores diagnostiques ont montré des corrélations significatives avec divers types de cellules immunitaires, à savoir les cellules T CD8, les cellules T. gamma. delta, les monocytes, les macrophages M0, les cellules dendritiques au repos, les cellules dendritiques d’activation et les neutrophiles dans la cohorte GSE26378 (Figure 4A), et B. cell. naïve, cellule T CD8, cellule T folliculaire auxiliaire, cellule NK d’activation, macrophage M1, cellule dendritique au repos et neutrophile dans la cohorte GSE13904 (Figure 4B), et de cellules T. folliculaires. auxiliaires, de cellules dendritiques. au repos et de neutrophiles dans la cohorte GSE26440 (Figure 4C). Les cellules dendritiques et les neutrophiles au repos présentaient une corrélation avec les scores du modèle dans les trois ensembles de données (Figure 4D). En raison de la présence limitée de cellules dendritiques au repos dans le sang périphérique, nous avons exclusivement confirmé la corrélation entre les neutrophiles et les scores du modèle dans des échantillons cliniques, ce qui a donné des résultats cohérents avec l’analyse susmentionnée (Figure 4E). L’intégration de ces résultats permet de déduire que la présence partagée de neutrophiles constitue potentiellement le mécanisme sous-jacent par lequel le gène du modèle de diagnostic influence l’apparition de la septicémie pédiatrique.
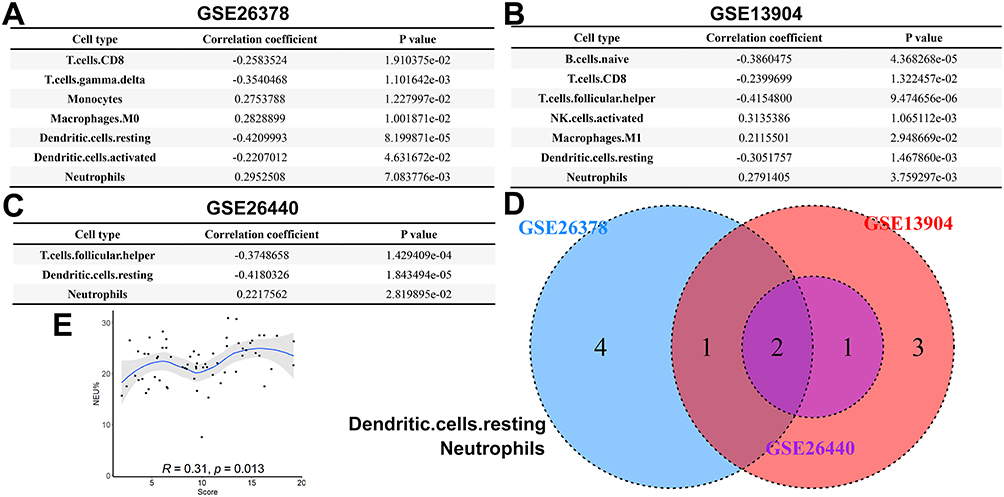 |
Figure 4 Analyse de l’infiltration des cellules immunitaires dans la septicémie pédiatrique. (A) Types de cellules associés aux scores des modèles dans la cohorte GSE26378. (B) Types de cellules associés aux scores des modèles dans la cohorte GSE13904. (C) Types de cellules associés aux scores des modèles dans la cohorte GSE26440. (D) Graphique de Veen de l’analyse de corrélation dans les trois cohortes. (E) La corrélation entre les neutrophiles et les scores de modèle dans les échantillons cliniques. |
Analyse d’enrichissement fonctionnel
Nous avons divisé les échantillons de septicémie pédiatrique en deux groupes, l’un élevé et l’autre faible, en fonction des scores diagnostiques et nous avons analysé les gènes différentiels entre les deux groupes en vue d’un enrichissement fonctionnel. Comme le montrent les Figure 5AL’analyse GO a montré que les gènes différentiels étaient principalement enrichis dans les sous-unités ribosomiques et les processus biologiques liés à la production et au métabolisme de l’ARN, tandis que l’analyse KEGG a montré que les gènes différentiels étaient principalement enrichis dans les processus biologiques liés aux lysosomes, aux ribosomes et au complément (Figure 5B).
 |
Figure 5 Analyse d’enrichissement fonctionnel. (A) Analyse du go. (B) Analyse KEGG. |
Discussion
Le sepsis pédiatrique, caractérisé par un dérèglement de la réponse immunitaire aux agents pathogènes, est une affection qui entraîne une morbidité et une mortalité importantes dans le monde entier.13 L’identification et la prise en charge opportunes du sepsis pédiatrique posent des défis considérables en raison des manifestations cliniques souvent variables qu’il présente. Les biomarqueurs susmentionnés, à savoir la CRP et la PCT, sont largement accessibles dans la majorité des établissements pédiatriques et continuent de servir d’outils cliniques précieux pour diagnostiquer le sepsis pédiatrique.19 Néanmoins, leur sensibilité et leur spécificité sont relativement limitées, ce qui rend nécessaire l’identification de nouveaux biomarqueurs capables de diagnostiquer rapidement et précisément les septicémies pédiatriques dans des contextes médicaux réels. Les biomarqueurs diagnostiques des lysosomes qui ont été développés dans notre étude présentent un potentiel considérable pour relever les défis cliniques susmentionnés. Cette approche diagnostique surpasse les diagnostics de biomarqueurs conventionnels et fait preuve d’une précision accrue dans la capture de la dynamique complexe du système immunitaire humain.
L’intégration du big data, de l’apprentissage automatique et de l’intelligence artificielle offre la possibilité de développer de meilleurs outils d’identification du sepsis, qui peuvent soutenir efficacement la prise de décision dans les cas de sepsis pédiatrique et faciliter la mise en œuvre d’approches thérapeutiques basées sur la précision.2 En utilisant des techniques d’apprentissage automatique pour l’identification des gènes cruciaux et en effectuant une analyse complète des données de séquençage à haut débit dérivées de multiples cas de septicémie pédiatrique, nous avons développé avec succès un modèle de diagnostic dans la présente étude. Ce modèle est capable de dépister efficacement le sepsis pédiatrique, comme le montre sa discrimination précise entre les cas normaux et les cas de sepsis pédiatrique, à la fois dans les échantillons cliniques de sang périphérique que nous avons collectés et dans les ensembles de données publiquement disponibles sur le sepsis pédiatrique. En outre, l’examen de la corrélation entre le modèle de diagnostic susmentionné et l’infiltration de cellules immunitaires a révélé une forte association. Cela peut être attribué à l’implication substantielle de la régulation immunitaire cellulaire, qui englobe les cellules immunitaires, dans la pathogenèse de la septicémie pédiatrique. Les monocytes et les macrophages jouent un rôle important dans l’inflammation et après l’attaque d’un agent pathogène.20 Les monocytes transforment la septicémie : de cellules dotées d’une puissante capacité immunomodulatrice positive, comme le Pha des bactéries, elles deviennent des cellules défectueuses incapables de mettre en place une défense efficace contre l’infection.13 Une éventuelle augmentation de la charge lysosomale au cours de la septicémie peut résulter d’une suppression de l’absorption bactérienne et de la dégradation lysosomale par les monocytes ou les macrophages.21 Les bactéries qui se cachent dans les lysosomes utilisent des monocytes ou des macrophages dysfonctionnels comme hôtes pour éviter l’activité normale des cellules immunitaires et les cytokines.13 En outre, la phagocytose est étroitement liée aux lysosomes dans la pathogenèse des maladies inflammatoires.8 La phagocytose et le trafic lysosomal sont nécessaires à la réponse inflammatoire des cytokines au peptidoglycane de Bacillus anthracis.22 Le traitement de la septicémie peut être entravé par des monocytes/macrophages dysfonctionnels, qui peuvent même aider les germes à échapper à la clairance immunitaire.13 Par conséquent, ces résultats ont le potentiel d’améliorer notre compréhension des mécanismes immunologiques moléculaires sous-jacents qui précipitent l’apparition de la septicémie pédiatrique.
Malgré l’utilisation de diverses données de séquençage à haut débit provenant du sepsis pédiatrique et l’application de techniques d’apprentissage automatique pour identifier les gènes caractéristiques, notre étude présente des avantages distincts par rapport aux enquêtes précédentes.17,23,24 Premièrement, notre modèle de diagnostic présente une efficacité diagnostique accrue, comme le montre son application réussie à des échantillons cliniques et sa validation ultérieure à l’aide de trois ensembles de données accessibles au public. Deuxièmement, le modèle de diagnostic que nous avons mis au point permet de faire la distinction entre les étiologies bactériennes et non bactériennes de la septicémie pédiatrique (AUC=0,82).
Notre étude comporte plusieurs limites. Premièrement, le manque de données sur la mortalité dans les trois ensembles de données sur le sepsis pédiatrique accessibles au public entrave l’évaluation de l’efficacité prédictive de notre modèle. Deuxièmement, l’influence potentielle des conditions comorbides chez les patients atteints de septicémie pédiatrique inclus dans notre recherche peut avoir un impact sur la précision de notre modèle dans la prédiction des résultats. Enfin, malgré les performances satisfaisantes du modèle que nous avons construit en analysant les 65 échantillons cliniques collectés, la taille limitée de l’échantillon et la nature monocentrique de l’étude nécessitent la conduite d’enquêtes multicentriques supplémentaires avec des échantillons plus importants pour valider et améliorer l’efficacité diagnostique du modèle.
Conclusions
Nous avons réussi à identifier un modèle de diagnostic composé de quatre gènes associés aux lysosomes, qui présente un haut niveau de fiabilité dans l’identification de la septicémie pédiatrique. Ce modèle présente des gènes candidats potentiels pour les tests de diagnostic utilisant des échantillons de sang périphérique chez les patients atteints de septicémie pédiatrique, et offre également de nouvelles perspectives pour le développement de thérapies ciblant les lysosomes dans cette population spécifique de patients.
Abréviation
GEO, gene expression omnibus ; ROC, Receiver operating characteristic curve ; DEGs, differentially expressed genes ; NETs, Neutrophil extracellular traps (NETs) ; CatB, cathepsin B ; GSEA, gene set enrichment analysis ; LASSO, Least absolute shrink and selection operator ; RF, random forests ; AUC, aire sous la courbe ; qRT-PCR, PCR quantitative en temps réel ; PCT, procalcitonine ; NEU%, pourcentage de neutrophiles ; WBC, globules blancs ; CRP, protéine C-réactive ; PBMCs, cellules mononucléaires du sang périphérique ; TRIPOD, Transparent Reporting of a multivariable prediction model for Individual Prognosis Or Diagnosis (rapport transparent d’un modèle de prédiction multivariable pour un pronostic ou un diagnostic individuel).
Déclaration de partage des données
Les ensembles de données utilisés et/ou analysés dans le cadre de la présente étude sont disponibles auprès de l’auteur correspondant sur demande raisonnable.
Approbation éthique et consentement à la participation
Cette étude a été soutenue par les comités d’éthique du premier hôpital affilié de l’université de Zhengzhou (2023-KY-0932-002). Le consentement éclairé écrit des parents ou des tuteurs légaux de tous les patients et des témoins sains a été obtenu. Toutes les méthodes ont été appliquées conformément aux directives et réglementations en vigueur. Le manuscrit est conforme à la déclaration d’Helsinki.
Contributions des auteurs
Tous les auteurs ont apporté une contribution substantielle à la conception, à l’acquisition des données ou à l’analyse et à l’interprétation des données ; ils ont participé à la rédaction de l’article ou l’ont révisé de manière critique pour en dégager le contenu intellectuel important ; ils ont accepté de le soumettre à la revue actuelle ; ils ont donné leur approbation finale à la version à publier ; et ils acceptent d’être responsables de tous les aspects de leur travail.
Financement
Cette étude a été financée par le projet de construction conjointe de la Commission provinciale de la santé et de la santé du Henan (LHGJ20230209).
Divulgation
Tous les auteurs ne déclarent aucun conflit d’intérêts dans le cadre de ce travail.
Références
1. Qin Y, Caldino Bohn RI, Sriram A, et al. Refining empiric subgroups of pediatric sepsis using machine-learning techniques on observational data. Front Pediatr. 2023;11:1035576. doi:10.3389/fped.2023.1035576
2. Singer M, Deutschman CS, Seymour CW, et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA. 2016;315(8):801–810. doi:10.1001/jama.2016.0287
3. Evans L, Rhodes A, Alhazzani W, et al. Surviving sepsis campaign : international guidelines for management of sepsis and septic shock 2021. Crit Care Med. 2021;49(11):e1063–e1143. doi:10.1097/CCM.0000000000005337
4. Wang F, Gómez-Sintes R, Boya P. Perméabilisation de la membrane lysosomale et mort cellulaire. Trafic. 2018;19(12):918–931. doi:10.1111/tra.12613
5. Yambire KF, Rostosky C, Watanabe T, et al. L’acidification lysosomale altérée déclenche une carence en fer et une inflammation in vivo. eLife. 2019;8. doi:10.7554/eLife.51031
6. Boya P, Kroemer G. Perméabilisation de la membrane lysosomale dans la mort cellulaire. Oncogene. 2008;27(50):6434–6451. doi:10.1038/onc.2008.310
7. Biasizzo M, Kopitar-Jerala N. Interaction entre l’inflammasome NLRP3 et l’autophagie. Front Immunol. 2020;11:591803. doi:10.3389/fimmu.2020.591803
8. Chen L, Zhao Y, Lai D, et al. Neutrophil extracellular traps promote macrophage pyroptosis in sepsis. Cell Death Dis. 2018;9(6):597. doi:10.1038/s41419-018-0538-5
9. Tsujimoto K, Jo T, Nagira D, et al. The lysosomal ragulator complex activates NLRP3 inflammasome in vivo via HDAC6. EMBO J. 2023;42(1):e111389. doi:10.15252/embj.2022111389
10. Yang R, Zhang X. A potential new pathway for heparin treatment of sepsis-induced lung injury : inhibition of pulmonary endothelial cell pyroptosis by blocking hMGB1-LPS-induced caspase-11 activation. Front Cell Infect Microbiol. 2022;12:984835. doi:10.3389/fcimb.2022.984835
11. Peng J, Pan J, Wang H, Mo J, Lan L, Peng Y. Morphine-induced microglial immunosuppression via l’activation de la mitophagie insuffisante régulée par NLRX1. J Neuroinflammation. 2022;19(1):87. doi:10.1186/s12974-022-02453-7
12. Liu Y, Wang D, Li T, et al. Melatonin : a potential adjuvant therapy for septic myopathy. Biomed Pharmacother. 2023;158:114209. doi:10.1016/j.biopha.2022.114209
13. Zhao Q, Gong Z, Wang J, et al. A zinc- and calcium-rich lysosomal nanoreactor rescues monocyte/macrophage dysfunction under sepsis. Adv. Sci. 2023;10(6):e2205097. doi:10.1002/advs.202205097
14. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis : a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–15550. doi:10.1073/pnas.0506580102
15. Weiss SL, Peters MJ, Alhazzani W, et al. Surviving sepsis campaign international guidelines for the management of septic shock and sepsis-associated organ dysfunction in children. Intensive Care Med. 2020;46(Suppl 1):10-67. doi:10.1007/s00134-019-05878-6
16. Zhang G. Regulatory T-cells-related signature for identifying a prognostic subtype of hepatocellular carcinoma with an exhausted tumor microenvironment. Front Immunol. 2022;13:975762. doi:10.3389/fimmu.2022.975762
17. Fan J, Shi S, Qiu Y, Liu M, Shu Q. Analysis of signature genes and association with immune cells infiltration in pediatric septic shock. Front Immunol. 2022;13:1056750. doi:10.3389/fimmu.2022.1056750
18. Gentles AJ, Newman AM, Liu CL, et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat Med. 2015;21(8):938–945. doi:10.1038/nm.3909
19. Lim PPC, Bondarev DJ, Edwards AM, Hoyen CM, Macias CG. The evolving value of older biomarkers in the clinical diagnosis of pediatric sepsis. Pediatr Res. 2023;93(4):789–796. doi:10.1038/s41390-022-02190-w
20. Germic N, Frangez Z, Yousefi S, Simon HU. Regulation of the innate immune system by autophagy : monocytes, macrophages, dendritic cells and antigen presentation (Régulation du système immunitaire inné par l’autophagie : monocytes, macrophages, cellules dendritiques et présentation des antigènes). Cell Death Differ. 2019;26(4):715–727. doi:10.1038/s41418-019-0297-6
21. Cai J, Li J, Zhou Y, et al. Staphylococcus aureus facilite sa survie dans les macrophages bovins en bloquant le flux autophagique. J Cell Mol Med. 2020;24(6):3460–3468. doi:10.1111/jcmm.15027
22. Iyer JK, Khurana T, Langer M, et al. La réponse des cytokines inflammatoires au peptidoglycane de Bacillus anthracis nécessite une phagocytose et un trafic lysosomal. Infect Immun. 2010;78(6):2418–2428. doi:10.1128/IAI.00170-10
23. Wang X, Guo Z, Wang Z, et al. Diagnostic and predictive values of pyroptosis-related genes in sepsis. Front Immunol. 2023;14:1105399. doi:10.3389/fimmu.2023.1105399
24. Zhang WY, Chen ZH, An XX, et al. Analysis and validation of diagnostic biomarkers and immune cell infiltration characteristics in pediatric sepsis by integrating bioinformatics and machine learning. World J Pediatr. 2023;19(11):1094–1103. doi:10.1007/s12519-023-00717-7
Source de l’article